文献研读|林国亮(2017级博士):Nat. Methods|等位基因组组装算法hifiasm
文献摘要:等位基因组(haplotype-resolved genome,亦即能分辨单倍型的基因组)是指组装出等位基因的基因组1–3,对于二倍体及以上倍性的物种而言,因为存在两套或两套以上的染色体组,存在不同的等位位点而存在不同的单倍型,因而组装出高质量的等位基因组是开展精细的基因组变异研究的必备基础。针对高度杂合的二倍体基因组在单倍体基因组组装中存在的遗传信息丢失等问题,作者基于PacBio HiFi reads,开发了等位基因组组装算法hifiasm。hifiasm利用HiFi reads高准确率的特性,构建定相string graph以保留单倍型信息。区别于其他组装算法仅保留一个完整的单倍型信息,hifiasm算法保留所有的单倍型信息。在3个人类和5个非人类的测试数据中,hifiasm 生成的等位基因组组装结果始终优于其他的等位基因组组装算法。同时,利用hifiasm算法提供完整的单倍型信息,作者开发的图形trio binning 算法(graph-based trio binning algorithm)优于标准的trio binning 算法(standard trio binning)。
研究背景:
等位基因组组装是研究基因组变异最理想的方式。但现有的算法中大多数忽略二倍体生物中的两个等位基因之间的差异,组装出一个嵌合的单倍体基因组(haploid genome)2,4,这种方法对于近乎纯合的近交样本非常有效,但对于高度杂合的二倍体基因组来说,一半的遗传信息可能会在此过程中丢失,使得研究人员很难对基因组中高度杂合区域进行深入的研究。
为了解决这个问题,一些组装算法陆续被发表(WhatsHap、Falcon-Unzip、HapCUT2等),这些算法都是包含如下步骤,第一,生成具有代表性的组装结构,第二,再据此生成一对包含有单倍型信息的组装结果(含完整的单倍型组装结果和由单倍型contig群的组装结果)。此外,近期发表的一些组装算法TrioCanu、HiCanu和FALCON-Phase等)通过尝试用额外的信息,如家系(father-mother-offspring trio)和Hi-C数据等,先将待组装测序reads划分到不同单倍型,再对不同单倍型分别进行组装,从而获得高质量的等位基因组组装结果。但是这些方法可能无法鉴别出复杂区域的单倍型相关markers,无法针对高度复杂的区域进行组装,从而导致组装错误。
为此,李恒团队开发出一种基于PacBio HiFi reads的等位基因组组装算法,能够有效地对大型复杂的基因组生成高质量的等位基因组组装结果。
hifiasm算法
hifiasm主要为三步:序列校正,定相 string graph构建和基因组组装(图1)。
序列校正:hifiasm采用all-vs-all比对的方法对HiFi reads进行纠错。基于reads之间的overlap信息,如果read的一个位点上包含的两种碱基比对上了,且每一种碱基都至少有3个以上的reads支持,则认为它是SNP并保留,否则认为是错误并进行纠正。hifiasm只使用相同单倍型的数据进行校正,从而避免过度校正。
定相string graph构建:基于第一步校正后的reads构建定相string graph,在定相string graph中,reads为顶点,重叠区为边。杂合位点信息通过气泡 “bubble” 的形式形成多条路径从而保留下来。
基因组组装:如果没有亲本的测序数据,hifiasm 会随机选择气泡的一边输出类似Falcon unzip和HiCanu的主要组装结果(primary contigs)。如果提供亲本的测序数据,hifiasm则可以通过亲本特有的k-mer在图上标记自父母本的序列(trio binning),从而得到等位基因组。
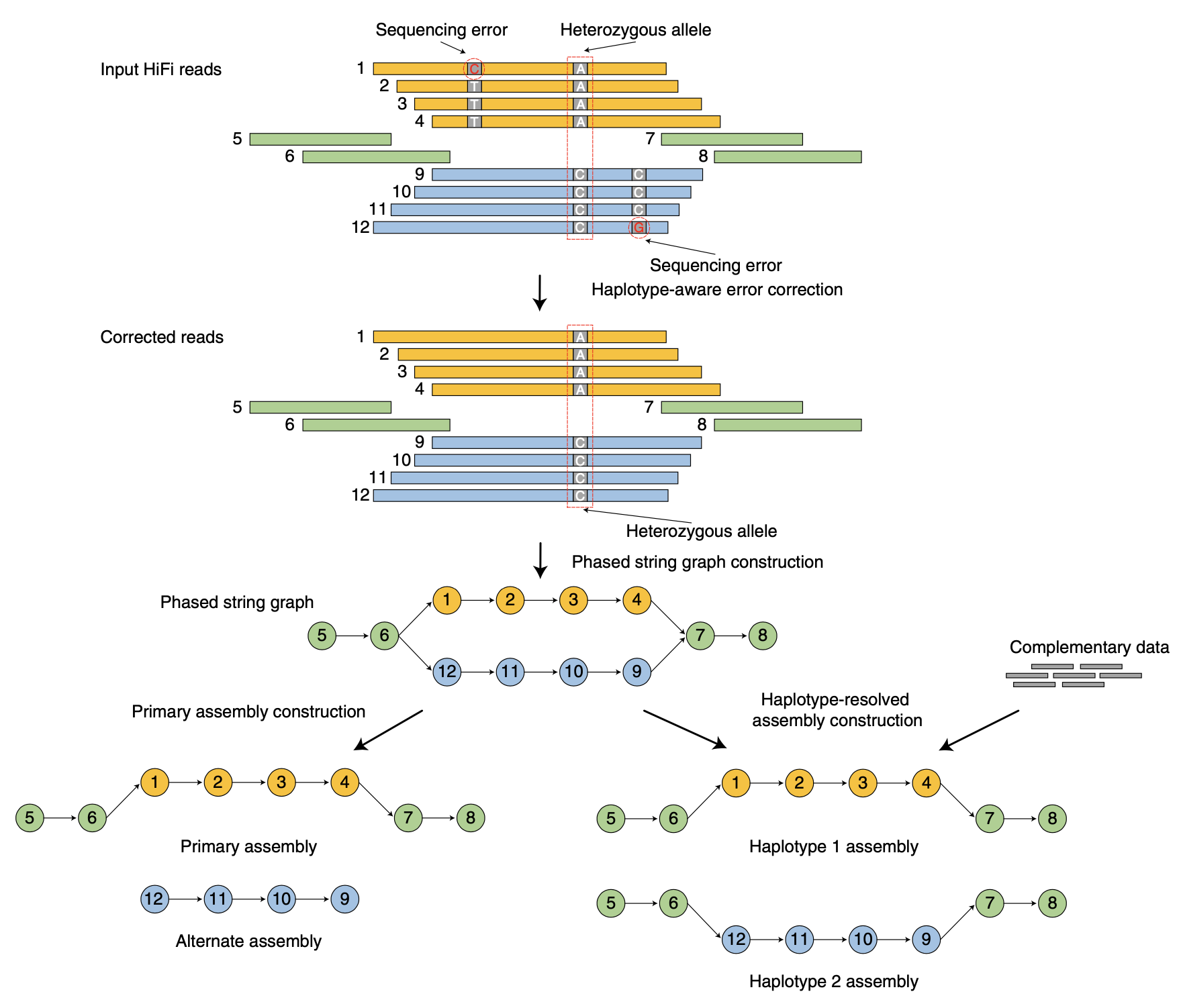
Fig. 1 | Outline of the hifiasm algorithm.
图1. Hifiasm算法组装流程图
基于标准的trio binning策略进行等位基因组组装的时候会存在如下问题:若双亲中至少有一方在某个位点上杂合,可能会导致reads不能被划分到具体的单倍型,这些不能划分的杂合reads在两个亲本的组装中都会被用到,从而导致错误。而hifiasm 则采用graph-based trio binning策略,它不会预先对reads进行划分,而是在string graph 标记reads。因此在很长的杂合信息位点“bubble”中,即使只有少数部分reads被标记,hifiasm也能很好的将其定相(图2)。
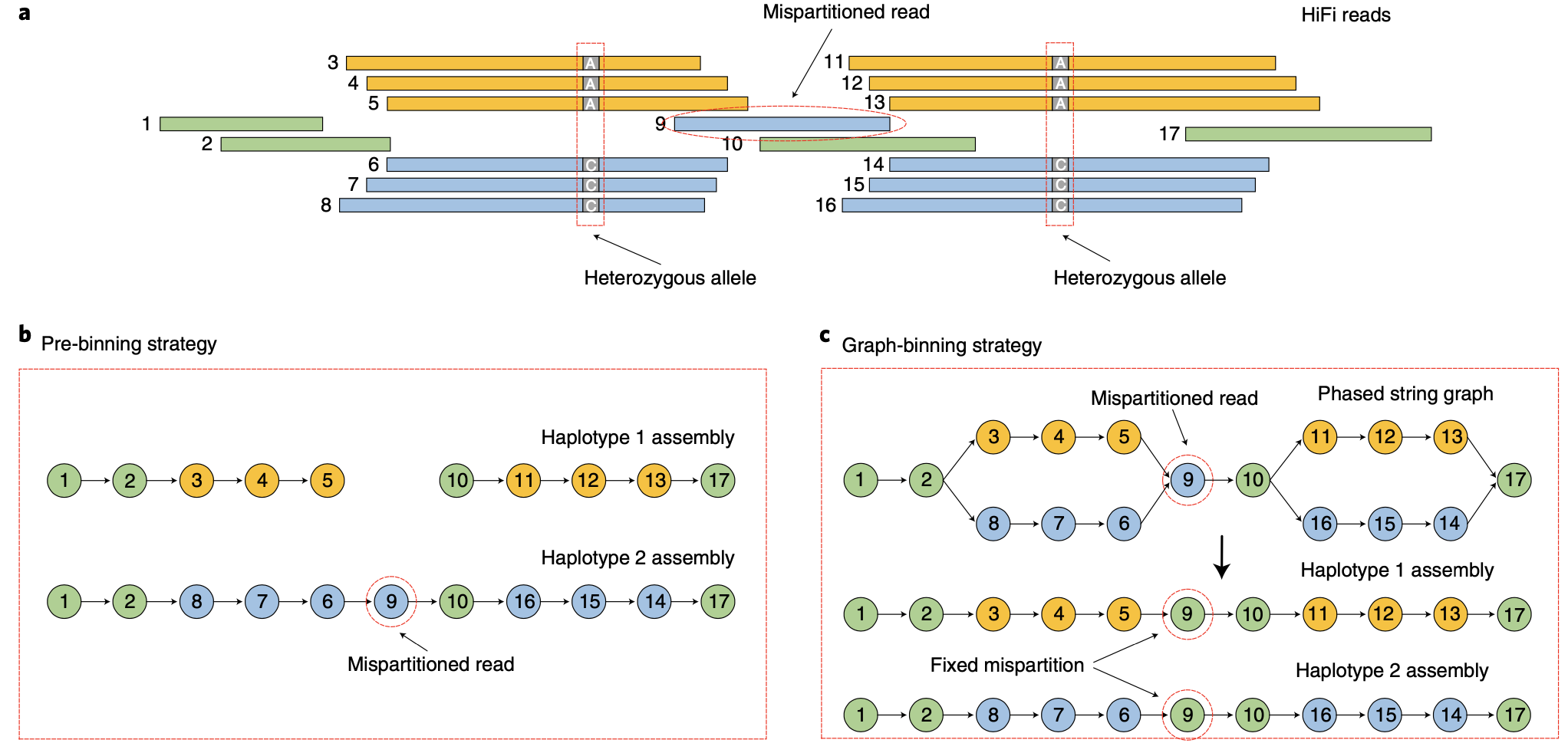
Fig. 2 | Effect of false read binning.
图2 错误划分reads对于组装等位基因组的影响
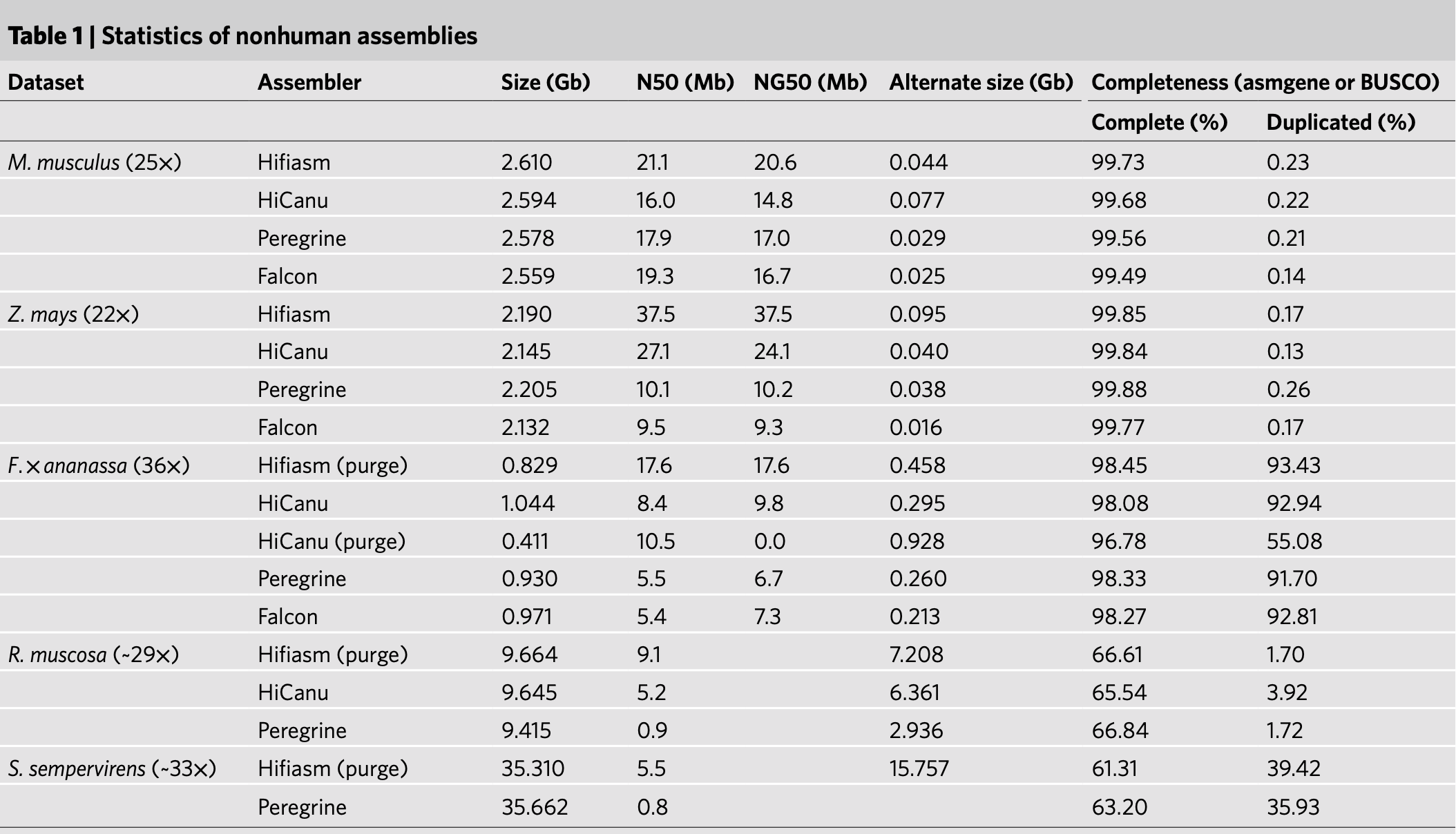
在完成对hifiasm算法的调试后,作者通过不同的数据集,测试了hifiasm、Falcon-Unzip、Peregrine和HiCanu这四款算法,并对算法性能及结果准确性进行了比较分析。作者首先在小鼠(Mus musculus)和玉米(Zea mays)的近交品系中测试发现,hifiasm和HiCanu组装结果优于其他两款算法(表1)。其次,作者又测试杂合度较高的草莓(Fragaria×ananassa), 黄腿山蛙(Rana muscosa)和六倍体加州红杉(Sequoia sempervirens)等基因组,比较结果显示,hifiasm都能够产生质量最高的组装结果且效率较高,尤其是在组装六倍体时,hifiasm仅用3天就完成了组装,而传统的Peregrine花费了15天,且hifiasm组装出来的序列N50是Peregrine 7倍(表1)。
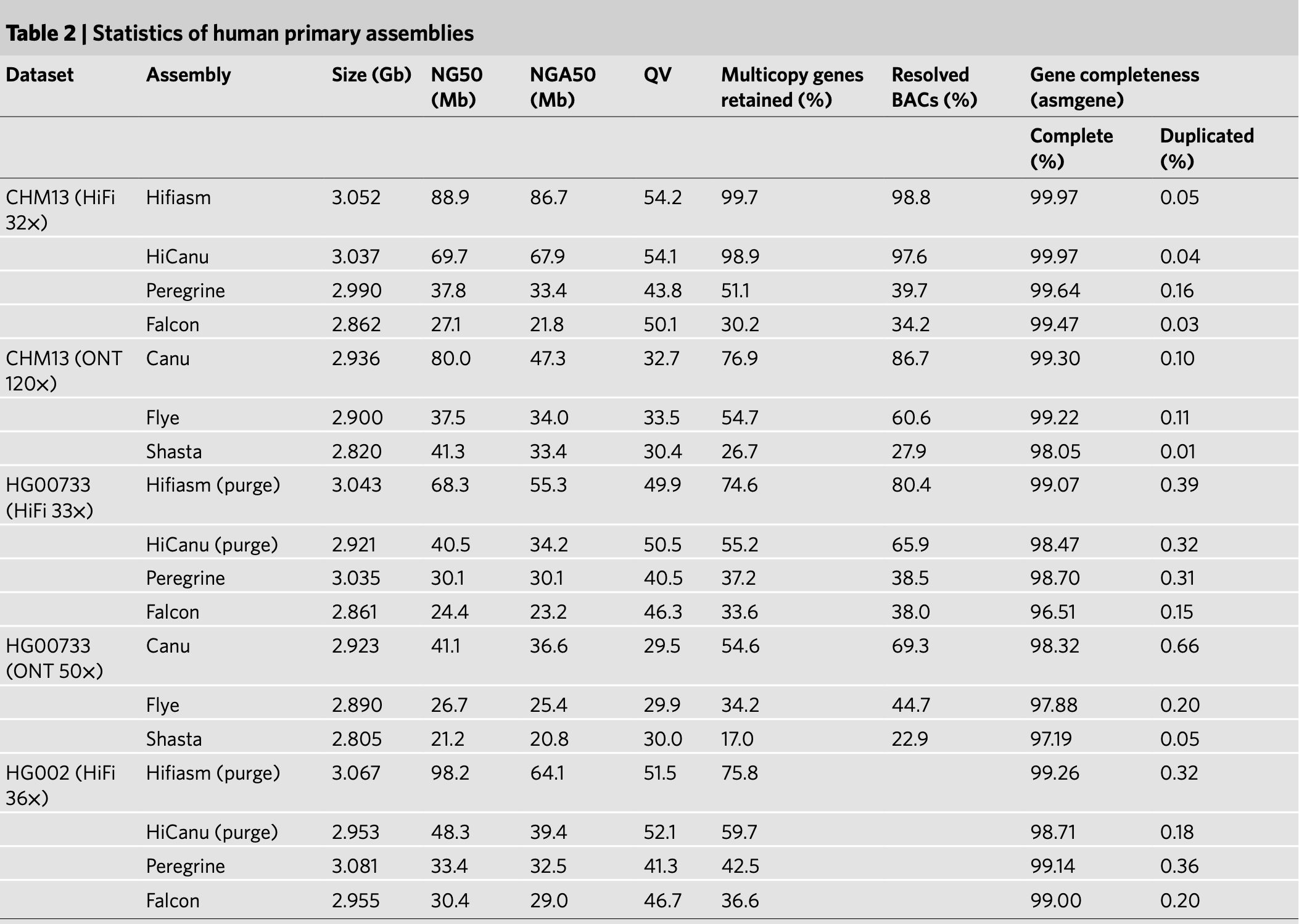
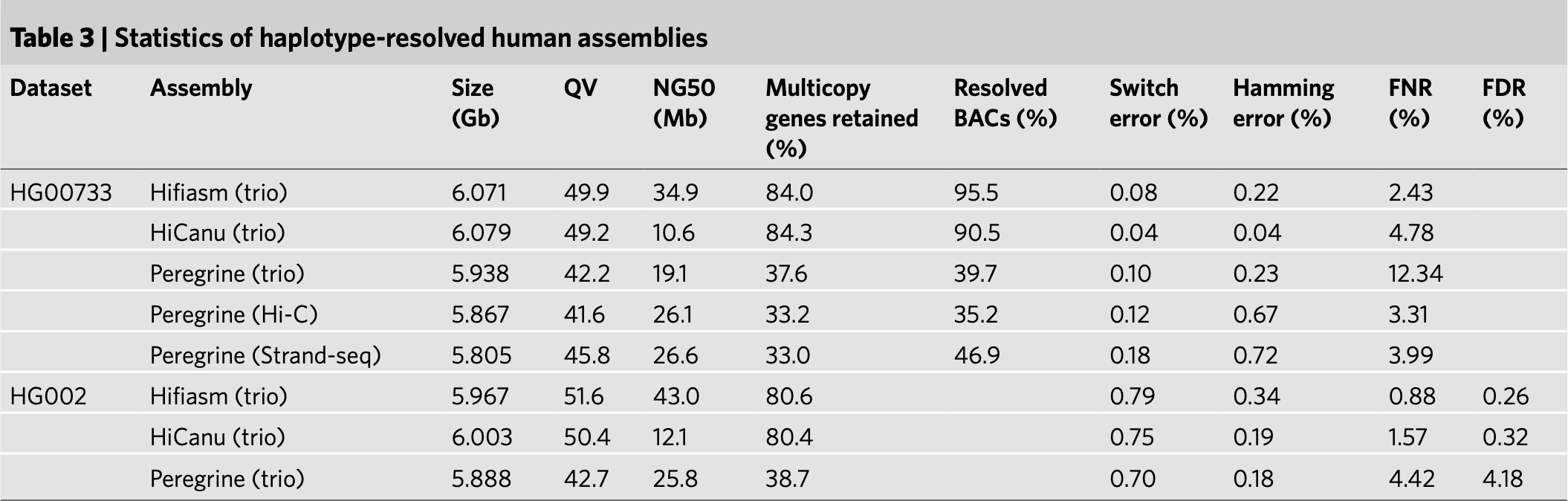
在人类基因组组装中,作者研究发现,无论是纯合的细胞系还是杂合的二倍体样本,hifiasm组装出来的基因组NG50值在比较的几款算法中最大,同时hifiasm也能够相对准确组装出多拷贝基因(表2),尤其是在基于trio binning策略组装等位基因组结果中,hifiasm 采用了graph-based trio binning 策略,相比于其他软件采用传统的trio binning 策略,hifiasm组装出来的NG50长度是其他算法的3倍以上(表3)。如对于具有高杂合度的人类白细胞抗原区,hifiasm 可以准确将其准确组装出来,而其他算法则无法准确组装。
总结
综上,hifiasm 是一个利用HiFi reads的进行从头组装等位基因组组装的算法。该算法利用HiFi reads构建定相的string graph保留了全部单倍型信息,这使得hifiasm可以组装出几乎一致的重复区域,获得的等位基因组组装结果准确,并且组装速度快。与其他算法相比,hifiasm性能最优算法。构建等位基因组有助于我们更好地研究单碱基突变和结构变异对基因调控以及表型的影响,可以预见,等位基因组组装将成为人类和动植物研究中的通用流程。综上,hifiasm解决了复杂重复区域组装难的问题,为快速准确组装等位基因组提供了便利,具有广泛的应用潜力。
研究者信息
本文由美国哈佛大学医学院Dana-Farber癌症研究所李恒团队研究发表,该团队长期致力于开发序列比对、序列拼装和寻找突变等一系列算法。该团队在全基因组测序,Hi-C和单细胞数据分析方面拥有非常丰富的专业知识和经验,并将这些计算技能应用于癌症基因组学数据,以提高对点突变和结构突变识别的准确性。
相关知识简述
hifiasm算法中用到的trio binning 技术是指利用父本、母本和子代的遗传信息对子代的单倍型划分的方法(图3),由美国国家人类基因组研究所、Pacific Biosciences公司及阿德莱德大学等单位的研究人员在2018年联合开发(Koren et al. 2018 Nat. Biotechnol.)。该方法的有效性随着杂合度的增加而提高,极大地提升了等位基因组的组装质量。如在Yang 等人利用该方法组装的狨猴等位基因组中,杂合性位点数量是之前的10倍,为狨猴的演化及生物医学研究提供了更加精确的等位基因组(Yang et al. 2021 Nature)。
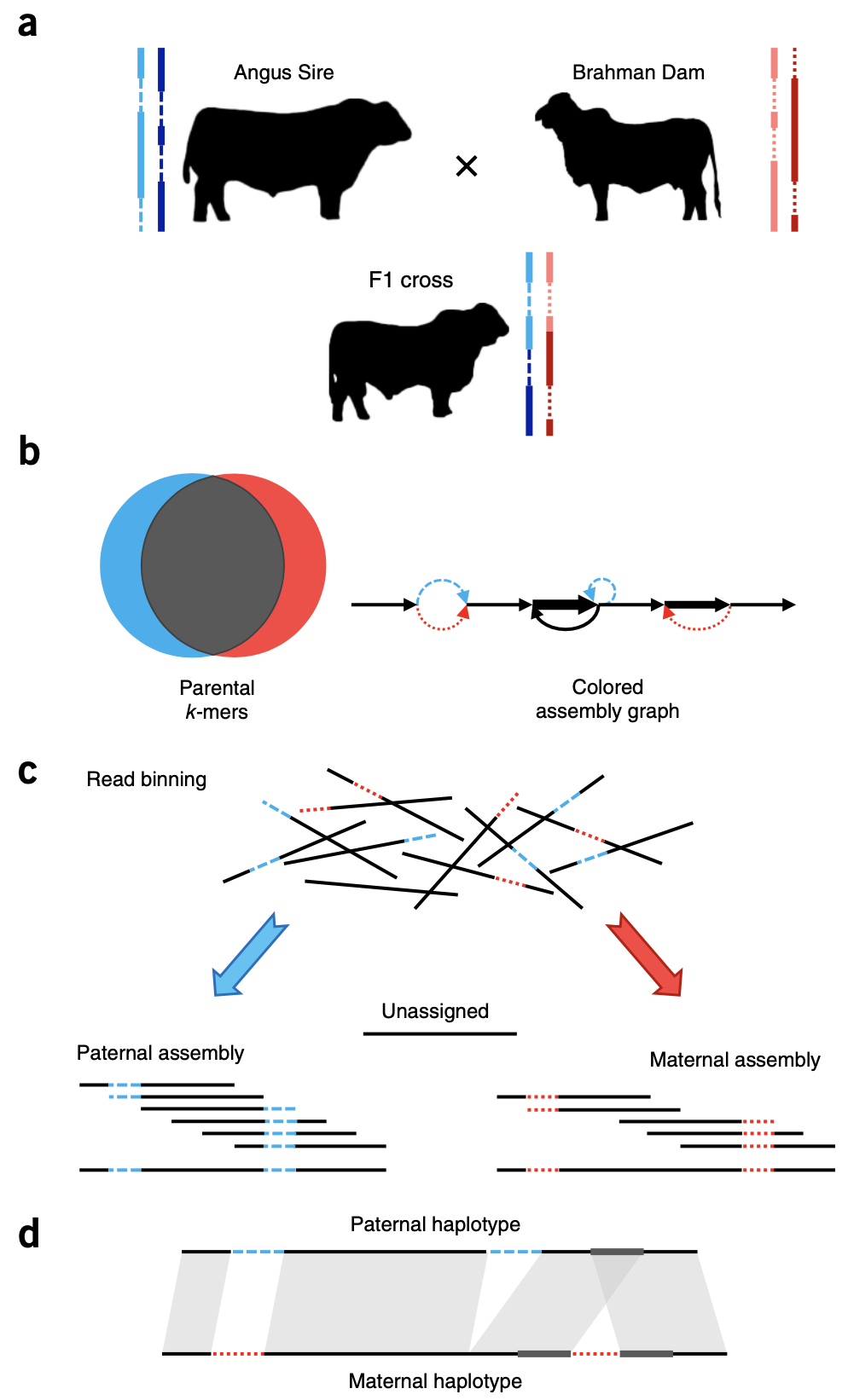
Fig 3| Outline of trio binning and haplotype assembly.
图3. 基于家系单倍型组装策略
原文传递:https://www.nature.com/articles/s41592-020-01056-5
此文献解读引用的其他参考文献:
1. Cao, H. et al. De novo assembly of a haplotype-resolved human genome. Nature Biotechnology 33, 617–622 (2015).
2. Garg, S. et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nature Biotechnology 1–4 (2020) doi:10.1038/s41587-020-0711-0.
3. Bansal, V., Halpern, A. L., Axelrod, N. & Bafna, V. An MCMC algorithm for haplotype assembly from whole-genome sequence data. Genome Res. 18, 1336–1346 (2008).
4. Kronenberg, Z. N. et al. Extended haplotype phasing of de novo genome assemblies with FALCON-Phase. bioRxiv 327064 (2019) doi:10.1101/327064.


